
Overview
Christine Seidman, MD and Jonathan Seidman, PhD have discovered genetic etiologies and mechanisms for human heart disease. Their work has provided fundamental insights into myocyte biology, enabled gene-based diagnosis, and defined novel therapeutic targets.
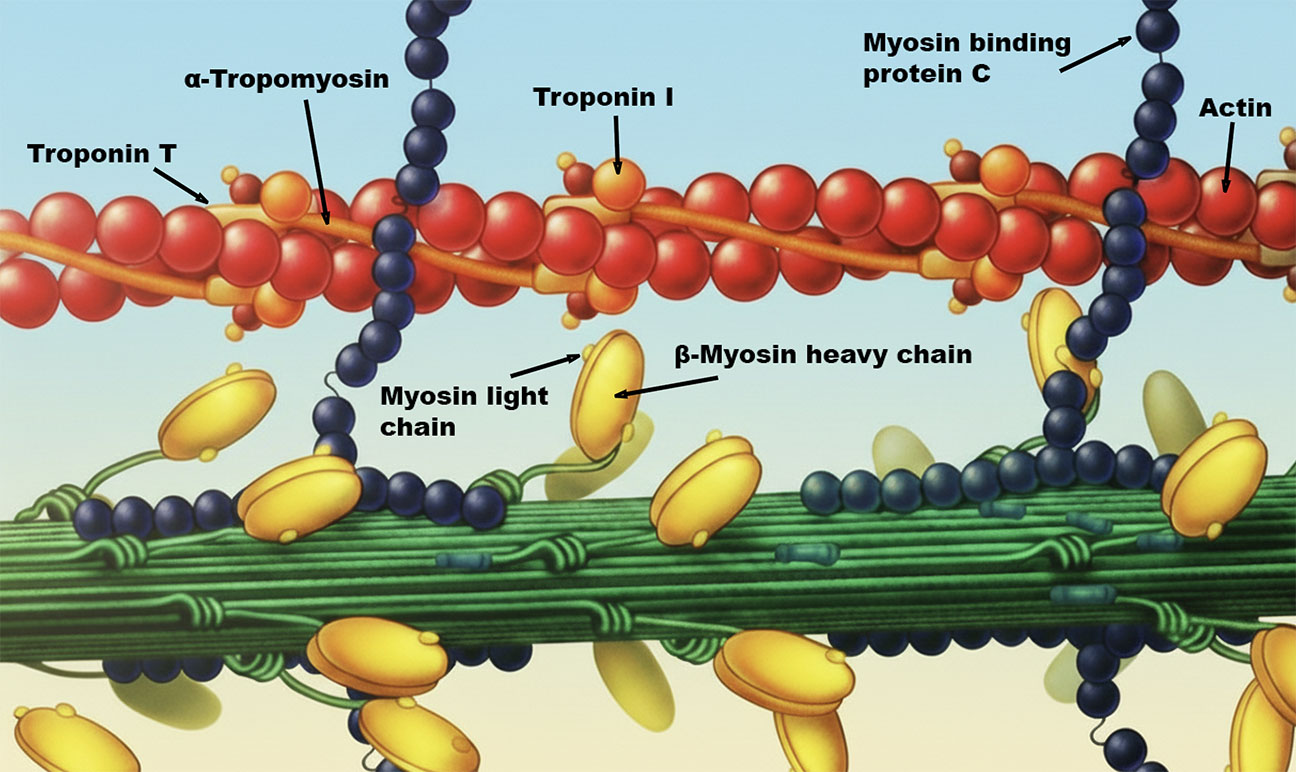
Hypertrophic cardiomyopathy (HCM) is a primary myocardial disorder characterized by unexplained increases in left ventricular wall thickness. HCM causes arrhythmias, stroke, and heart failure and is the most common cause of sudden death in athletes and of non-violent sudden death among young adults. The Seidmans showed that HCM is caused by dominant mutations (usually missense) in genes encoding sarcomere proteins including myosin, myosin binding protein C, troponin T and I, and a-tropomyosin. Through analyses of HCM mouse models they demonstrated that mutations increase sarcomere power by amplifying acto-myosin sliding velocity, force generation, and ATP hydrolysis. The increased biophysical properties of the sarcomere activate TGF-beta signaling, which promotes myocardial fibrosis and diminishes cardiac relaxation, events that can be inhibited by antibody-blockade or angiotensin II (Type 1) receptor antagonists. These pathways explain the increased cardiac contractility and diastolic dysfunction that characterizes HCM, led to the demonstration of increased circulating levels of profibrotic molecules in HCM patients, and provide the rationale for ongoing clinical trials to assess the potential benefit of TGF-beta inhibition. To more directly ameliorate this genetic disease, the Seidmans recently developed shRNAs that selectively silences HCM transcripts, and showed the long-term in vivo efficacy, including suppression of all cardinal manifestations of disease, in mice carrying HCM mutations.
Review: Hypertrophic cardiomyopathy: Translating cellular cross talk into therapeutics
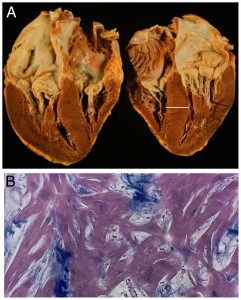
Dilated cardiomyopathy (DCM) describes an enlarged and weakened myocardium that often culminates in heart failure. DCM can arise as a primary heart muscle disease, a scenario often attributed to unrecognized infection or toxins. The Seidmans unended this paradigm, showing that gene mutations in lamin A/C, eyes-absent-4, phospholamban, myosin heavy chain, troponin T, and titin cause unexplained DCM. The impact of titin mutations in DCM is substantial. Truncating titin mutations (nonsense, frameshift, or splicing) are the most cause of DCM occurring approximately 20% of familial and sporadic cases, more than all other DCM genes combined. Surprisingly, titin truncating mutations are non-randomly distributed across this 33,000 amino acid peptide that spans half (~0.5 m) of the sarcomere. These are over-represented in the carboxyl A-band domain, an observation that implies a dominant negative mechanism. By modeling DCM gene mutations in mice, the Seidmans have dissected the molecular responses and explained the variable clinical manifestations and outcomes observed in DCM patients, efforts that have improve the precision of therapeutic interventions.
Review(s): Genetic causes of human heart failure
Truncations of Titin Causing Dilated Cardiomyopathy
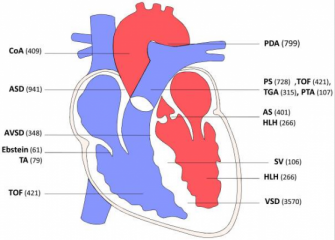
The Seidmans discovered the first dominant mutations in cardiac transcription factors (TBX5 and NKX2.5) that cause congenital heart defects (CHD). The developmental programs perturbed by these mutations in mouse models provide fundamental insights into myocyte differentiation pathways and as well explained electrophysiologic defects in CHD patients. By applying advanced genomic strategies, they discovered de novo copy number variants and mutations in histone modifying genes were identified in severe CHD. Remarkably, these genes and loci overlap with genetic causes of neurocognitive disorders, perhaps explaining the frequent co-occurrence of these disorders in patients.
Review: Genetics of Congenital Heart Disease: The Glass Half Empty